Precisely defining disease variant effects in CRISPR-edited single cells
Jul 23, 2025·
 ,,,,,,,,,,
,,,,,,,,,,
Yuriy Baglaenko
Corresponding, equal contribution
Zepeng Mu
Equal contribution
,Michelle Curtis
Hafsa M Mire
Vidyashree Jayanthi
Majd Al Suqri
Cassidy Liu
Ryan Agnew
Aparna Nathan
Annelise Yoo Mah-Som
David R Liu
Gregory a Newby
Soumya Raychaudhuri
Corresponding
·
0 min read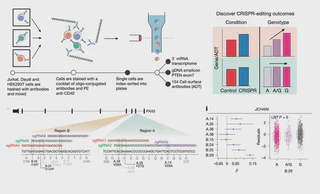
Abstract
Genetic studies have identified thousands of individual disease-associated non-coding alleles, but the identification of the causal alleles and their functions remains a critical bottleneck. CRISPR–Cas editing has enabled targeted modification of DNA to introduce and test disease alleles. However, the combination of inefficient editing, heterogeneous editing outcomes in individual cells and nonspecific transcriptional changes caused by editing and culturing conditions limits the ability to detect the functional consequences of disease alleles. To overcome these challenges, we present a multi-omic single-cell sequencing approach that directly identifies genomic DNA edits, assays the transcriptome and measures cell-surface protein expression. We apply this approach to investigate the effects of gene disruption, deletions in regulatory regions, non-coding single-nucleotide polymorphism alleles and multiplexed editing. We identify the effects of individual single-nucleotide polymorphisms, including the state-specific effects of an IL2RA autoimmune variant in primary human T cells. Multimodal functional genomic single-cell assays, including DNA sequencing, enable the identification of causal variation in primary human cells and bridge a crucial gap in our understanding of complex human diseases. A plate-based assay called CRAFTseq has been developed that uses ‘multi-omic’ single-cell RNA sequencing and direct genotyping of CRISPR edits to test the functional effects of genetic variants on cell state and function.
Type
Publication
Nature